金币
UID743052
帖子
主题
积分4764
注册时间2021-9-13
最后登录1970-1-1
听众
性别保密
|
欢迎您注册蒲公英
您需要 登录 才可以下载或查看,没有帐号?立即注册
x
01发补
Q:(上海+注册+华艳):CDE技术审评的发补是需要等到综合审评才有发补通知书吗?注册检验的发补也是综合审评阶段吗(注册检验有某项有问题没检,单项需要再次注册检验)?
A:(宁波-注册-小月):技术审评发补是综合审评后。注册检验通知书会在注册受理时发给企业,CDE好像很少对注册检验有发补意见吧,通常在给技术发补意见的时候,会要求对变更项再次进行标准复核,或者复核检验。跟技术审评意见一起提的。如果发补时标准中增加项,需要针对该检测项进行再次注册检验。
02校正因子
Q:(深圳-注册-李开):1%自身对照乘以校正因子法中杂质校正因子:如果杂质限度是0.1%。标准曲线法测定校正因子,其中:主成分的线性是以0.1%为100%还是以1%为100%?
A:以主成分的0.1%为100%(尽量主成分取点百分比跟杂质一致,这样计算的校正因子误差小),因为原料未知杂质的限度是0.10%,因此限度的100%是0.1%。
03公司名称变更
Q:参比制剂一次性进口批件中公司名称发生了变更,可以走变更手续吗?
A1:(上海~化药注册~张杰):实物药品上面的公司名称能跟已发批件对得上么?对的上就可以用。
Q:(豫小南):药品没问题,就是申请批件的公司名称变更了。参比制剂能有A公司购买赠送给B行不行?
A1:(上海~化药注册~张杰):委外机构可以,我们申请人和研发机构之间有委托合同就行。
A2:可以,写个情况说明,将变更前后的公司关系及参比制剂购买情况说明清楚就可以。
04稳定性数据
Q:(上海-注册-星星):现在IND申报1个月的稳定性数据可以吗?
A1:(Frank-Yang-RA-上海):看你临床使用周期,需要覆盖使用周期。
A2:建议能够提供3个月的稳定性数据,3个月基本能支持开展I期临床了,前提还是覆盖使用周期。
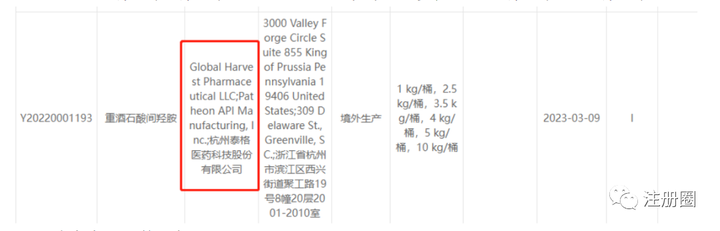
05授权书
Q:(注册er):第一个和第二个企业名称不同的,如果开授权书,应该是第一个开还是第二个开呢?
 [backcolor=rgba(18, 18, 18, 0.5)]​ [backcolor=rgba(18, 18, 18, 0.5)]​
编辑切换为居中
添加图片注释,不超过 140 字(可选)
A1:(沪-打杂-���):第一个。
Q:(注册er):所以不同的时候都是默认持证人开授权书?
A2:(上海-冯新宇-RA):一般是这样,如果第一家实在开不了,COA是谁家出的就开谁家的,开不了得再出关系声明。
06申报资料
Q:(绿意):化学药品申报资料的3.2.A附录需要提供吗?
A1:(注册er):不需要。
A2:如果有药典未收载的非新型辅料,建议放3.2.A.3。
Q:(绿意):那3.3的参考文献需要单独搞一份文件吗?
A1:(注册er):有参考文献就统一放3.3。
07稳定性研究
Q:(北京-注册-四叶草):这种情况30℃加速考察几个月呢?
 [backcolor=rgba(18, 18, 18, 0.5)]​ [backcolor=rgba(18, 18, 18, 0.5)]​
编辑切换为居中
添加图片注释,不超过 140 字(可选)
A:(沪-打杂-���):注册时接受提交只到6个月的数据。
08一致性评价
Q:(豫小南):口服固体制剂的一致性评价申报还是按照之前的120号文整理资料吗?也是需要按现在的电子申报吗?
A1:(北京-注册-媛媛):固体制剂是的,液体制剂是国家药监局药审中心关于发布《化学药品注射剂仿制药质量和疗效一致性评价技术要求》等3个文件的通告(2020年第2号)。
A2:(上海-注册-贰雯):按照《国家药监局关于实施药品注册申请电子申报的公告》(2022年第110号)要求。
 [backcolor=rgba(18, 18, 18, 0.5)]​ [backcolor=rgba(18, 18, 18, 0.5)]​
编辑切换为居中
添加图片注释,不超过 140 字(可选)
09制剂申报
Q:(长春-研发注册-YXL):报制剂的时候要交原料药的方法学资料吗?
A1:需要,可以放在S部分。
A2:看你以什么方式递交,如果原料药已经单独登记了,就不需要了。
10CDE临床登记
Q:(上海-RA):CDE临床登记可以授权给CRO的账户进行登记,这种状态vs企业账户/子账户登记的区别是什么?如果选择CRO的账户进行登记,是否方便企业主账户随时做一些操作,比如“取消”。
A:主账户可以看到所有的信息,子账户看到的关键信息甚少,还有就是子账户只能看到自己子账户上提交的一些文件,其他子账户上的信息看不到。不管是谁的账户进行登记, 责任主体都是最终的申请人,用这种方式并不能转移责任主体,因此企业主账户不能随时做一些操作,比如“取消”。
11申请人主体变更
Q:临床试验获批后,要把项目转给其他公司,想要变更申请人主体,请问是否需要向CDE提交变更申请呢?
A:(馨文):不需要,只需要合同。
12药品注册检验
Q:(长春—药品注册—罗):药品注册检验需要提交注册标准的起草说明么?
A:(侦察连最差的兵):需要起草说明和质量控制部分研究资料。
13CEP申请
Q:申请CEP提供的GPS坐标都是用什么测的呢,手机自带的指南针只能显示4位小数,要求是提供5位小数。
A:(浙江-注册-April):手机显示度分秒,换算到度小数点5位。
14微生物检测
Q:(长春—药品注册—罗):注册检验原液的最小包装用1.5ml的EP管,但原液要检测微生物限度,检测微生物的项用50ml的储液袋装是否可以?
A1:(注册圈-苏州-江小白):包材不一样了,不可以的。可以用大点的同材质包材。
15原料药年报
Q:(北京-注册-宏艳):请问大家有准备原料药年报递交的吗?在CDE申请人之窗递交还是在NMPA药品年度报告采集模块递交呢?2022年16号文(国家药监局关于印发《药品年度报告管理规定》的通知),附件2模版适用于原料药吗?
A:原料药的年报在CDE申请人之窗递交,16号文主要针对的是制剂,原料药可以参考2019年56号文。
16电子签章
Q:(小燕):请教大家,备案申请表使用电子签章,持有人和生产企业不是同一家,电子签章日期能使用PDF编辑加上去吗?
A:(左手倒影):红色电子签章软件直接就可以签日期。
17 IND申请
Q:(Arti):IND申请这里是否只提供安全性评价机构证明文件,其他委托研究机构的需要说么?
 [backcolor=rgba(18, 18, 18, 0.5)]​ [backcolor=rgba(18, 18, 18, 0.5)]​
编辑切换为居中
添加图片注释,不超过 140 字(可选)
A:(张欢):一般提供非临床研究安全性评价机构的GLP批准证明或检查报告等证明性文件,临床试验机构应提供备案证明。
18勘误申请
Q:(云):原料药批件的附件需要勘误,这个勘误申请是寄给CDE业务管理处还是国家局行政受理服务大厅呢?
A:(樱花):电子版红头文件发给CDE,可以通过申请人之窗递交公文。
19杂质控制
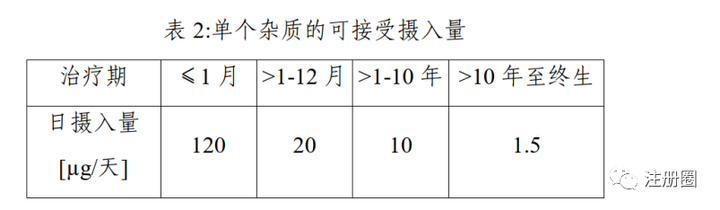
Q:(Alice):ICH M7(R1)明确“以与DNA反应的化合物甲磺酸乙酯为例(参考文献6,7),其阈值已通过体外和体内的致突变性建立。在阈值已确定的情况下,可以使用不确定因子计算PDE,代替线性外推。”我们通过查阅文献6,7得到甲磺酸乙酯大鼠的阈值NOEL为25mg/kg,经计算PDE为104μg/天,甲磺酸乙酯限度这样制定,论据充分吗?
A1:(京-GMP-David):参考:M7 R1。
 [backcolor=rgba(18, 18, 18, 0.5)]​ [backcolor=rgba(18, 18, 18, 0.5)]​
编辑切换为居中
添加图片注释,不超过 140 字(可选)
A2:可以的。但是也要考虑行业的整体水平,不少企业在NDA或ANDA时都是按照1.5μg/天控制的。如果是IND,按照上表控制即可。
20共线风险评估
Q:(王晶):对于共线风险评估如何查询这个品种是否属于细胞毒性类产品?从哪里可以查询?
A:(melon):从作用机制来判断,是否破坏细胞的结构。
21补充申请
Q:(竹七):下面这几个表在临床期间补充申请时,需要提交吗?在这份文件“(2019年5月6日发布)关于提交药品注册检查检验用申报资料光盘的通知(CDE)”中提到的五个表。1.药品研制情况信息表2.药品生产情况信息表3.现场主文件清单4.药品注册临床研究试验信息表5.临床试验信息表。
A:(猫熊的眼睛):不需要。
22现场主文件
Q:(Chloe):现场主文件这个文件只是注册申报时需要吗,工厂本身的文件体系不需要吗?
A1:(pengzhen):要有,你注册里的内容最后都要转化或者都是有依据来源的。
A2:(京-GMP-David):SMF要按文件体系要求管理的,版本签批都应该有的,里面的各种图纸也应当是签批过的。
23关联审评
Q:(勺子):19年登记的原料药没有受理号,制剂可以被受理么?现在是不是所有关联审评的原料药都必须提供受理号?
A:(夏天):有登记号就行可以关联审评。
24注册分类
Q:(.):一产品原研4mg规格的产品已经进口到国内,并在销售,也已经有了仿制药。原研6mg规格的产品还没有进口,但已经有了仿制药。申报4mg是按4类报,这个6mg是按3类,还是可以按4类?
A:(乐一图):一起报可以按4类。多规格申报时候注册分类选4类就行了。
25BE试验
Q:(vivian):是否遇到过这样的品种:仿制品种,FDA要求做空腹和餐后2项BE,但国内只做一项就批准了的?
A:BE试验做空腹还是餐后,还是两个状态都做,需要根据说明书中用法用量,参考相关生物等效性指导原则,结合FDA 具体品种生物等效性指导原则而定;目前未了解到有“仿制品种,FDA要求做空腹和餐后2项BE,但国内只做一项就批准了的”这种情况。
26增加适应症
Q:(似水流年):1.目前进口药品国家局备案从备案受理到拿到更新后的证件需要多久,5+60吗?2.可以不可以同一个品种并行同时进行备案和补充申请,因为我们有一项是工艺变更中等变更,走备案类的话有可能会要求进行进行注册检验吗?
A:(1)如果是中等变更合并审批类变更,审评时限为六十日(不涉及现场核查的)。(2)不同类别变更同时进行时,按更高级别变更管理,不存在同时进行备案和补充申请。正常情况下走备案类的话不会要求进行进行注册检验。
27临床申报
Q:(似水流年):2.2类改良型制剂申报临床,原料为我公司自研,尚未完成工艺验证和稳定性6个月考察,可以采用原料中试批的资料和稳定性数据随制剂申报临床吗?等原料完成后续工作再单独申报?
A:可以用于临床试验申请。上市申请时需要原料完成后续工作,可以原料独立审评,也可以和制剂关联审评。
28说明书
Q:(山东-RA-CH):现在上市申请中,制定说明书时,要完全按22年发布的说明书撰写指南书写吗?药代动力学要放在【临床药理】项下吗?
A:需要。还有国家药监局药审中心关于发布《化学药品说明书及标签药学相关信息撰写指导原则(试行)》的通告(2023年第20号)。
29DS DP
Q:(Stemirna therapeutics-张军):DS和DP的区别究竟是什么呢?什么阶段划分的样品被称为真正的DS?
A:具体问题具体分析,没有明确规定,一般情况下DS为建立细胞库到无菌过滤液,DP为原液到成品。
30辅料登记
Q1:():辅料供应商不愿意在CDE登记,如果在我们制剂注册申请提供辅料的研究资料,需要哪些资料?
Q2:(CC):辅料不强制要求登记可与制剂申请一并提交目前还是征求意见稿么,有实际操作过的么?
A:(1)辅料的工艺研究、质量研究、稳定性研究、安全性研究等能证明这个辅料安全可靠、质量可控的相关研究资料。(2)目前还是征求意见稿。
31质量标准
Q:(YiLu):一个速释片剂允许存在两个有关物质检测方法吗?分别适用于不同供应商的原料。
A:审评不会有问题,只要你能证明你的方法科学合理,能控制产品质量。原料内控标准可以不同供应商用不同的方法,制剂不建议如此操作,一是方法学验证两个都要做,二是上市后生产检验效率会很低。
声明:本文转载来源于公众号【注册圈】,所有问答均来源于注册圈社群,非官方问答,仅供参考。
|
|



 |手机版|蒲公英|ouryao|蒲公英
( 京ICP备14042168号-1 ) 增值电信业务经营许可证编号:京B2-20243455 互联网药品信息服务资格证书编号:(京)-非经营性-2024-0033
|手机版|蒲公英|ouryao|蒲公英
( 京ICP备14042168号-1 ) 增值电信业务经营许可证编号:京B2-20243455 互联网药品信息服务资格证书编号:(京)-非经营性-2024-0033






 发表于 2023-3-27 10:42:50
发表于 2023-3-27 10:42:50
 置顶卡
置顶卡 变色卡
变色卡