欢迎您注册蒲公英
您需要 登录 才可以下载或查看,没有帐号?立即注册
x
对于临床试验用药品(Investigational Medicinal Product, IMP),企业通常无需开展完整的生产工艺验证。然而,对于非最终灭菌的无菌制剂,即使是临床试验用药品,也是需要开展除菌过滤工艺验证的。
gempex德恩咨询的GMP专家们在多次执行EU QP审计项目中发现,部分临床试验用药品的生产商存在未在临床阶段使用目标药液/药物开展除菌过滤工艺验证,甚至未充分评估滤芯供应商提供的研究/验证资料对目标产品的适用性等问题。企业通常面临以下现实挑战:
① 专业设备匮乏:缺乏开展细菌截留试验(Bacterial Retention Testing, BRT)的关键设备,如细菌截留装置。 (P.S.:图源自网络)
② 物料成本高昂:使用目标药液进行验证后,药液必须废弃。在临床阶段生产批量有限且价值高昂的情况下,验证的物料成本构成显著负担。
③ 工艺变更风险:临床阶段的工艺、配方或滤器供应商存在变更可能性。过早进行完整验证可能导致后续变更触发再验证的需求。
面对这些挑战,从除菌工艺验证的角度出发,临床试验用药品生产企业应该如何最大程度地满足监管合规性;更重要的是,保证非最终灭菌的无菌产品的质量以及患者的安全健康。本文旨在从GMP咨询公司的专业视角,解析相关法规要求,并提出应对策略,助力企业建立合规、高效、可执行的除菌过滤工艺验证。
01 相关法规指南要求
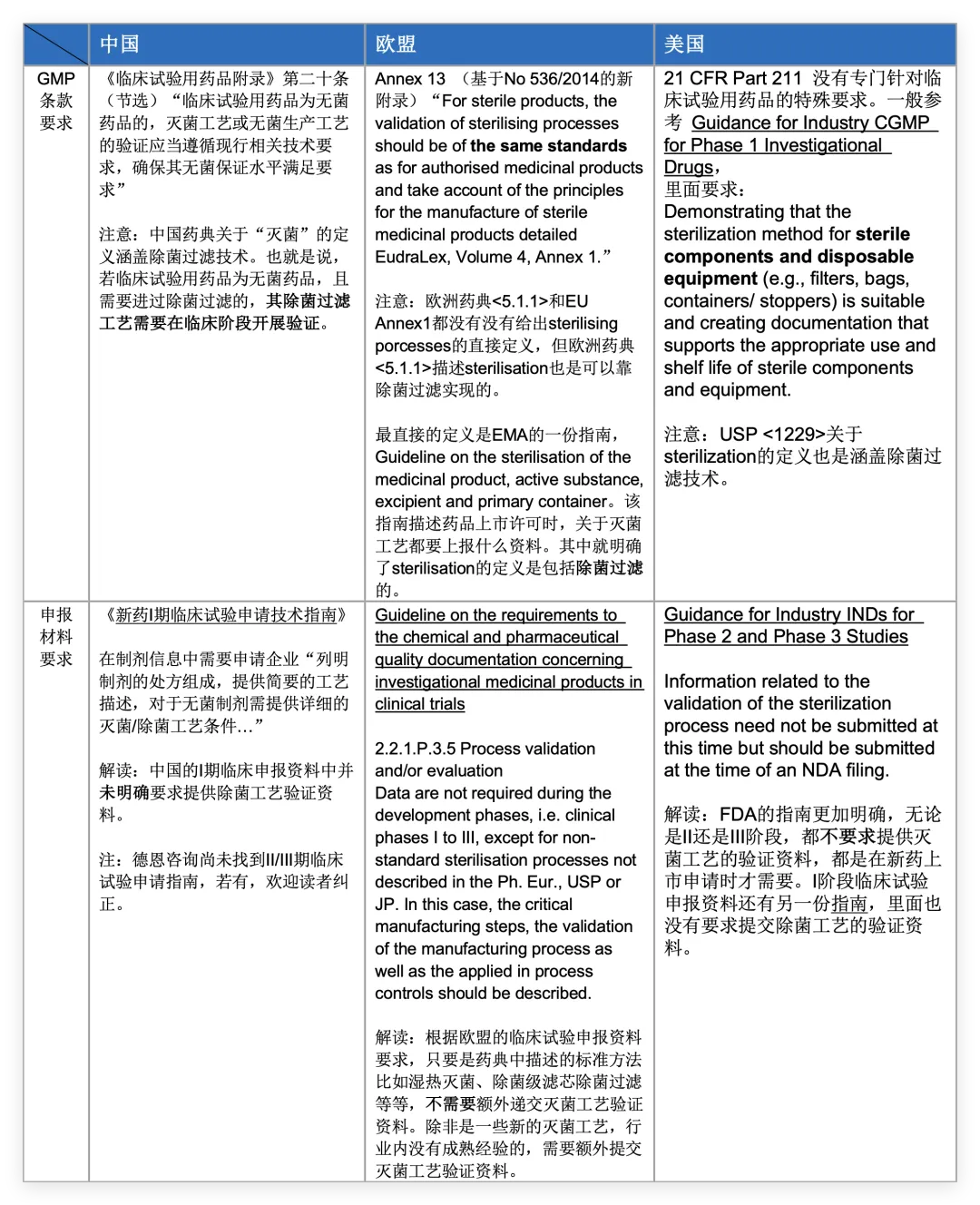
首先,确保合规的前提是深入理解监管机构的要求和期望。以下列出了中、美、欧三大主流市场关于临床阶段非最终灭菌制剂除菌过滤工艺验证的核心法规要求以及临床试验申报资料要求(节选)。 (点击图片即可放大查看)
小结: GMP条款或cGMP指南方面: - 中国与欧盟的GMP比较明确和严格,要求临床用药品生产用到的灭菌(除菌过滤)工艺要开展验证。
- 美国FDA则稍宽松,没有用到“验证”一词,药企能提供相应证据证明灭菌工艺的适用性就可以。
临床试验申报资料方面:三地监管机构在临床试验申报阶段均展现出相当的灵活性。对于采用药典载明的除菌过滤工艺,均未强制要求在临床申请资料中提交完整的工艺验证报告。核心要求在于工艺本身需满足GMP。
02 对于临床试验药企的实践建议
那么,临床试验用药生产企业应该要做到什么程度?德恩咨询觉得最保险的方法是用目标药液开展完整的除菌过滤工艺验证,但由于成本高、物料消耗大,还需具备相应设备和技术能力,往往在临床阶段做不到。另一种方法是,一些药企会拿到滤芯供应商提供的性能确认报告,然后基于此判定是否供应商所开展的研究结论是否适用于自己的制剂溶液。从法规指南来看,监管也是给了一定的灵活性,没有强制要求使用目标药液开展验证。比如:
1. EU Annex 1 8.84 During filter validation, wherever possible, the product to be filtered should be used for bacterial retention testing of the sterilising grade filter. Where the product to be filtered is not suitable for use in bacterial retention testing, a suitable surrogate product should be justified for use in the test. The challenge organism used in the bacterial retention test should be justified. 翻译: EU GMP 附录1第8.84条: 在过滤器验证期间,在可能的情况下,应当采用待过滤产品进行除菌级过滤器的细菌截留试验。若待过滤产品不适用于进行细菌截留试验,则应当证明选用某种替代产品进行该试验的合理性。细菌截留试验中所使用的挑战微生物应当予以论证其合理性。 2. 中国 新的无菌附录草稿版 第一百八十四条 应当尽可能将挑战微生物直接接种在待过滤产品中进行细菌截留试验。如果产品或工艺条件本身可能会影响挑战微生物的存活力,应当提供充足的数据和解释,证明替代产品的适用性和合理性。应当评估细菌截留试验的挑战微生物选择的合理性。 3. FDA Sterile Drug Products Produced by Aseptic Processing — Current Good Manufacturing Practice Direct inoculation into the drug formulation is the preferred method because it provides an assessment of the effect of drug product on the filter matrix and on the challenge organism. (.....) Any divergence from a simulation using the actual product and conditions of processing should be justified. 翻译: FDA《无菌工艺生产无菌药品——现行药品生产质量管理规范》:
"将挑战微生物直接接种于药品处方中进行试验是首选方法,因其可同步评估药品对滤膜基质及挑战微生物的影响。(......)任何偏离采用实际产品和工艺条件进行验证的方案,均需论证其合理性。"
小结: 基于上述法规要求,德恩咨询建议药企可考虑以下方面:
1. 最推荐的做法是用目前产品药液去开展除菌过滤工艺验证; 2. 若条件不允许,则至少需要充分评估滤器供应商的相关研究/验证资料是否涵盖药企目标产品的特性和工艺,包括但不限于以下方面: - 产品的pH值、极性、离子强度、黏度、表面张力、渗透压;
- 滤器的灭菌方法、除菌过滤工艺时间、温度、压力、过滤量与过滤膜面积之比等;
- 完整性检测的润湿液以及标准;
- 实验设计是否满足制药企业所声称的“冗余过滤”(若有)。
德恩咨询扎根GMP领域23年,已为全球企业执行过5000+项目。在药品的全生命周期中,我们团队可为企业量身定制除菌过滤工艺验证策略,还可提供从供应商第三方审计、差距分析、风险评估、验证方案设计与执行到检查支持的全面支持,助力企业化解合规挑战。
| 

 |手机版|蒲公英|ouryao|蒲公英
( 京ICP备14042168号-1 ) 增值电信业务经营许可证编号:京B2-20243455 互联网药品信息服务资格证书编号:(京)-非经营性-2024-0033
|手机版|蒲公英|ouryao|蒲公英
( 京ICP备14042168号-1 ) 增值电信业务经营许可证编号:京B2-20243455 互联网药品信息服务资格证书编号:(京)-非经营性-2024-0033





 发表于 2025-8-22 10:48:30
发表于 2025-8-22 10:48:30

 置顶卡
置顶卡 变色卡
变色卡