欢迎您注册蒲公英
您需要 登录 才可以下载或查看,没有帐号?立即注册
x
由于COVID-19新冠疫情影响,欧盟委员会于2020.03.25发布了寻求延迟一年实施MDR的提案,其目的在于减轻欧洲国家当局和工业界的压力,使其能集中精力应对和处理与冠状病毒危机有关的紧急优先事项。虽说MDR延迟一年,也就是2021年5月26日执行,目前部份公告机构已开始受理MDR认证申请。对此,整理了MDR法规下无菌器械审核关注点,供各位参考。 1 灭菌确认 当器械以无菌状态投放市场时,或预期由最终用户进行灭菌的器械,制造商应向最终用户提供灭菌参数和说明,并验证推荐的灭菌方法。因此,灭菌确认是公告机构审核的第一项重点。
相关标准和指南 1)EN 556-1 医疗器械的灭菌-被指定为“STERILE”的医疗器械的要求-第1部分:最终灭菌的医疗器械的要求; 2)ISO14937 医疗产品的灭菌-描述灭菌剂特征以及开发、确认和常规控制医疗器械灭菌过程的通用要求; 3)EN ISO 11135 医疗产品的灭菌-环氧乙烷-开发、验证和常规控制医疗器械灭菌过程的要地; 4)EN ISO 11137-1 医疗产品的灭菌-辐照-第1部分:开发、验证和常规控制医疗器械灭菌过程的要求; 5)EN ISO 11137-2医疗产品的灭菌-辐照-第2部分:确定灭菌剂量; 6)EN ISO 17665-1 医疗产品的灭菌-湿热-第1部分:医疗器械灭菌过程的开发、验证和常规控制的要求; 7)ISO 13408-1 医疗产品的无菌操作灭菌-第1部分:一般要求; 8)ISO 11138-1 医疗产品的灭菌-生物指示剂-第1部分:一般要求; 9)EN ISO 10993-7 医疗器械的生物学评估-第7部分:环氧乙烷残留; 10)AAMI TIR 28 环氧乙烷灭菌产品采用和过程等效。 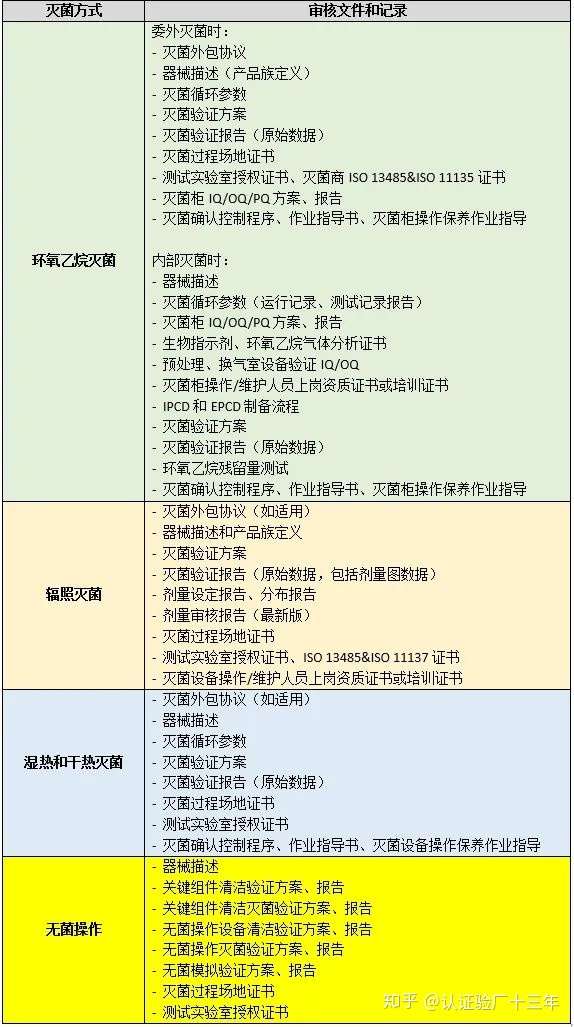 包装验证是审核的第二项重点。制造商应在声称的有效期内证明无菌医疗器械的产品和包装系统的稳定性,包含以下几个方面: 灭菌过程兼容性 包装对医疗器械无不良影响 与标签系统兼容性 物理、化学和微生物保护 包装材料不会浸出有害物质 保持无菌 运输和存储期间的保护
相关标准和指南 EN ISO 11607-1 最终灭菌医疗器械的包装-第1部分:材料、无菌屏障系统和包装系统的要求; EN ISO 11607-2 最终灭菌医疗器械的包装-第2部分:成型、密封和组装过程的验证。 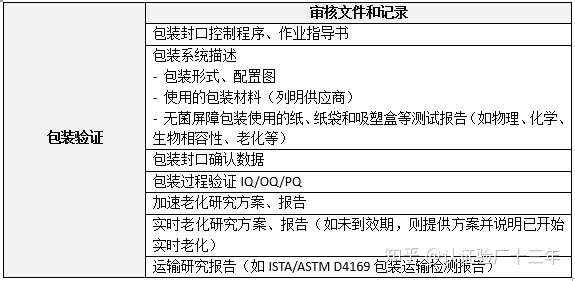 除此之外,公告机构还会评审无菌器械的生物相容性、风险管理、临床评估、标签和说明书等文件。 总而言之,技术文件需按照Regulation(EU) 2017/745 附录II和附录III编制,满足欧盟医疗器械MDR法规要求才可以通过公告机构的评审。 3 ENISO13485质量管理体系 医疗器械法规Regulation(EU) 2017/745中提到对于有CE标识的医疗器械制造商需要建立质量管理体系以确保批量生产的器械持续符合MDR法规的要求。同时建立上市后监督系统,并与所涉及器械的风险等级和类型相称。为了使风险最小化或防止发生不良事件,制造商应建立风险管理系统、事故报告和安全纠正措施的体系。 制造商应当建立、记录、实施和维护、更新并不断地完善质量管理体系,确保符合MDR法规第10条(9)的要求。 因此,质量管理体系的合规性是公告机构的另一项评审重点。 产品质量的控制是每一个企业头痛的问题,质量控制是一个系统工程,有其自身的规律和独特的控制方法;如果不掌握正确的质量控制方法,就很难控制产品的质量,甚至会出现一些意想不到的品质问题,给企业造成很大的经济损失。 但质量管控绝非易事,这是一个是企业的竞争力所在。 疫情当下,不论MDR法规是否延迟,为迎接这场硬仗企业提前做好充分的准备始终是不变的规划,正所谓以不变应万变,乃上上策。
| 

 |手机版|蒲公英|ouryao|蒲公英
( 京ICP备14042168号-1 ) 增值电信业务经营许可证编号:京B2-20243455 互联网药品信息服务资格证书编号:(京)-非经营性-2024-0033
|手机版|蒲公英|ouryao|蒲公英
( 京ICP备14042168号-1 ) 增值电信业务经营许可证编号:京B2-20243455 互联网药品信息服务资格证书编号:(京)-非经营性-2024-0033






 发表于 2021-2-5 10:28:40
发表于 2021-2-5 10:28:40
 置顶卡
置顶卡 变色卡
变色卡