金币
UID743052
帖子
主题
积分4789
注册时间2021-9-13
最后登录1970-1-1
听众
性别保密
|
欢迎您注册蒲公英
您需要 登录 才可以下载或查看,没有帐号?立即注册
x
01供应商备案
Q:(成都 注册 candy):原品种有两个原料药供应商,在一致性评价时只用了其中一家做工艺验证,现在一致性评价通过后还能不能直接用另一家供应商的原料药呢?还是只能再去做一次原料药增供的备案呢?
A:(***):增加备案。
02GMP条件
Q:(似水流年):IND申报,注册批一定要GMP条件下生产吗?
A1:(向野):临床样品符合GMP条件下生产是必要的(不一定要经过官方检查),因为你要保证临床用药安全,安全性、有效性、可靠性,你看哪个排在第一位,而且现在很多IND批产品都考虑用临床一期的,即使ind申报不查,后面也很麻烦。
A2:(pengzhen):没有法规说IND申报,注册批是GMP条件,很多企业中试是试验室做的,临床试验样品法规要求是GMP条件下生产的。当然你要是符合GMP条件肯定更好。
A3:IND申报,注册批不做临床,没有强行要求要在GMP条件下生产。
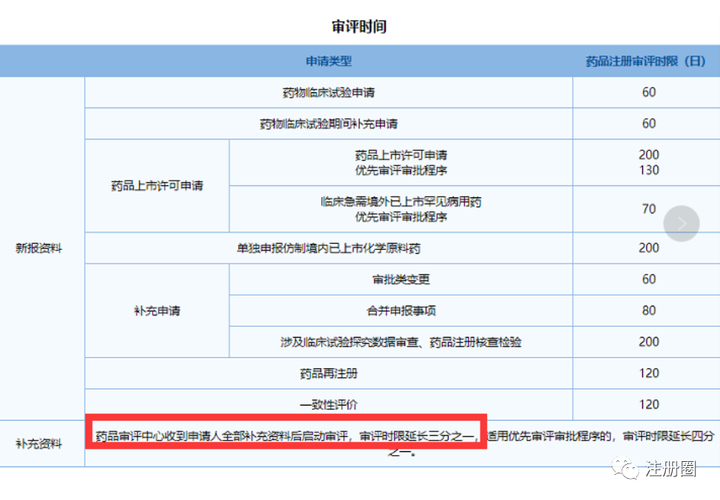
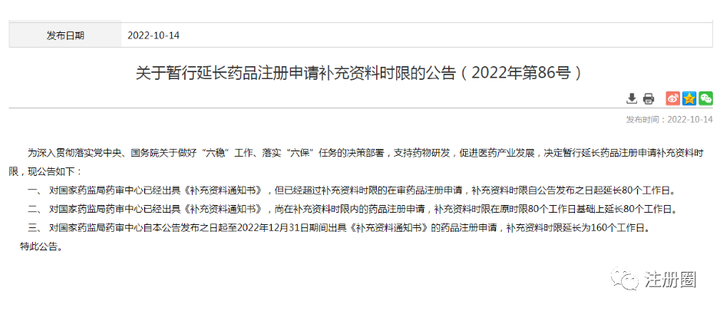
03补充资料时限
Q:(中山 生物制品研发 尹姑娘):补充资料时限的160个工作日应该不包含审评时限吧?这样算来,整个注册申请的时限就是 200个工作日+补充资料160个工作日+补充资料审评 200个工作日的三分之一,是这样理解吗?
 [backcolor=rgba(18, 18, 18, 0.5)]​ [backcolor=rgba(18, 18, 18, 0.5)]​
编辑切换为居中
添加图片注释,不超过 140 字(可选)
 [backcolor=rgba(18, 18, 18, 0.5)]​ [backcolor=rgba(18, 18, 18, 0.5)]​
编辑切换为居中
添加图片注释,不超过 140 字(可选)
A:(苏州 注册 冷了个冷):一共200+67(如果有发补)工作日。发补期间不计入审评时限,这个延长160工作日,耽误的是你自己的时间,不计在267工作日内。
04电子申报
Q:(Amber):新的电子申报要求说:实施电子申报前,申请人已提交药品注册申请且已受理的,审评过程中补充资料等仍采用纸质申报资料形式进行递交。对于新申请已经受理的,12月份还需要提交全套的纸质资料吗?电子申报实施前,电子和纸质的一起提交才算是受理是吗?
A:(Chloe):1月1日前受理的还是按照原来的方式走,光盘受理后提交纸版的。
05质量标准
Q:(上海 临床试验 Emily):工艺中使用了一个溶剂是三类溶剂,且多批次产品均未检测出,那么不体现在COA中可以接受吗?符合规范吗?
A:(苏州 注册 冷了个冷):正式批次多批次未检出,或低于限度10%,可以不定入标准和COA。
06关联审评
Q:就是我们申报临床的制剂产品的一个辅料生产企业,想跟我们做关联审评,该怎么做呢?是单独审评还是与制剂申请关联审评呢?
A:(启德医药RA):根据我的了解,临床阶段不关联审评的,就是先让他们提交到CDE的原辅包平台,拿到备案号,等有制剂品种用了他的辅料在NDA阶段的时候,CDE就去关联审评了。
07原料药批准
Q:(注册):现在原料药获批后发什么证书么?哪个文说的啊?
A1:(珠海 注册 Jason):国家药监局综合司公开征求《关于化学原料药再注册管理等有关事项的公告(征求意见稿)》意见:
 [backcolor=rgba(18, 18, 18, 0.5)]​ [backcolor=rgba(18, 18, 18, 0.5)]​
编辑切换为居中
添加图片注释,不超过 140 字(可选)
A2:2019年11月29日国家药监局发布的《关于贯彻实施《中华人民共和国药品管理法》有关事项的公告》(2019年第103号),明确2019年12月1日起,对化学原料药不再发放药品注册证书,由化学原料药生产企业在原辅包登记平台上登记,实行一并审评审批,所以现在获批的原料药没有批准文号,只有登记号,但获批后状态会由I变为A。
08供应商变更
Q:(广东-注册-王荣):活性炭(供注射用)厂家变更,大家觉得按什么变更处理?
A:活性炭在注射剂中主要起吸附热原、脱色、助滤等作用,活性炭(供注射用),需要评估不同厂家活性炭在注射剂除热原中的应用、对注射剂元素杂质的影响、对主药含量的影响、对注射剂不溶性微粒的影响。如果质量和技术等级不变,且此厂家此活性炭已关联申报过的话,按中等变更风险较小。
09杂质控制
Q:(上海 临床试验 Emily):原料药生产过程中所有使用的有机溶剂,是否都要进行控制、和残留溶剂检测?在COA中体现?
A:(苏州 注册 冷了个冷):这个属于杂质的控制,看你们自己内部控制策略和能力。如果很多批次都没有检测出就不需要。如果绝大部分批次检测出且和标准差不太多,那就要去进行控制。没有固定的要或不要的答案。
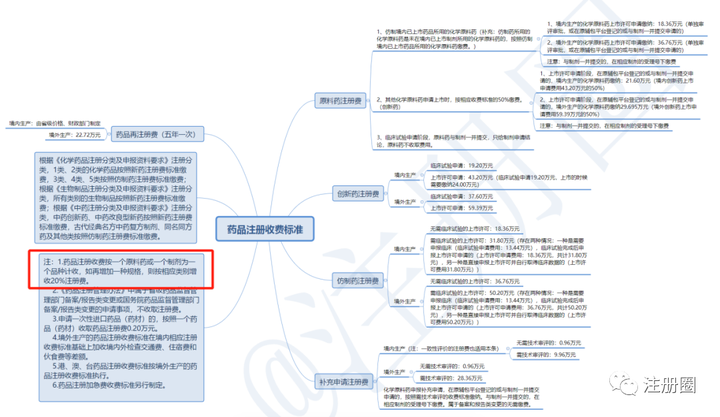
10注册费用
Q:(山东RAX):软膏或乳膏有多个包装规格,如15g、30g和60g,这种注册费用是按1个规格还是3个规格缴纳?
A:应该是这个。
 [backcolor=rgba(18, 18, 18, 0.5)]​ [backcolor=rgba(18, 18, 18, 0.5)]​
编辑切换为居中
添加图片注释,不超过 140 字(可选)
11原料药备案
Q:(Insider 庞):有个原料药备案问题,工艺验证前CPP有6个,验证完成后,在评估减少3个CPP,那过程控制里面描述是按照工艺验证描述6个,还是按照后来的3个?
A1:(长坡厚雪):您这个不是完成工艺验证了吗?应该将风险评估更新一下就行了吧,更新之后再按3个来进行描述。
A2:一般研发思路是,在验证前通过前期研究确定CPP,然后在验证阶段加以验证。建议还是按6个来吧,如果理由十分充分,也可以按3个来。如果审核的老师问起,要能够说服老师。
12稳定性考察
Q:(静小静):关于生物制品的有效期变更涉及的稳定性考察,是否需要进行正置及倒置条件下的稳定性考察,还是只需要正置就可以呢?
A1:(凡容):正置,倒置是做相容性的比较多,正常的稳定性考察不需要。具体可以看看药典规定。
A2:申报美国的话,需要考察最差放置条件。
13工艺前延
Q:(Sword):有没有遇到过NDA阶段需要工艺前延的情况的?如果工艺前延的生产厂是另外一个公司,而CDE又需要这个公司提供生产许可证,大家是如何处理这个问题的呢?
A:(注册圈):只能协助这个公司申请生产许可证。
14剂量变更
Q:(永恒梦想):现在有个项目参比是多剂量,我们研究按单剂量做BE申报,发补让变更为跟原研一样的多剂量包装,补充相应的药学和稳定性研究。最后会批准的只有多剂量,还是单剂量和多剂量一起批准?
A:(注册圈):大概率是多剂量。仿制药仿制的是原研!
15原料药备案
Q:(张爱萍):仿制没有原研进口,但国内已上市的原料药备案,是按3类,还是按4类提交资料。
A:(注册圈):按3类。
16方法学
Q:(tingY):我们一个复方制剂两个规格,工艺相同,处方比例有差异,做方法学时耐用性大规格的做了,小规格是否也得做?
A:这个需要根据你大规格和小规格选择的分析方法是否相同而评估,如果不同规格分析方法中样品浓度和杂质对照品浓度一致或没有显著差异,其他检测条件一致,可以不用做。
17项目转报
Q:(Lily):转报项目,就是自家产品已经在国外上市多年,现在转报国内,是什么程序呢?
A:(注册圈):不知道这里的自家产品具体是什么情况,自己拥有知识产权,在国外生产吗?如果该产品是在国外生产上市,贵司在国外持有,原研进口到国内按5.1类申报,仿制药进口到国内按5.2类申报。如果现在是国外生产上市,拟准备在国内生产并上市,如果国内无其他同品种上市,按3类;如果国内已有其他同品种上市,按4类进行申报.
18疫苗上市申请
Q:疫苗上市申请时,特性鉴定、结构确证应该怎么做?做哪些内容呢?
A:(注册圈):可以参考20200814CDE发布的《新型冠状病毒预防用疫苗研发技术指导原则(试行)》
1.1 灭活疫苗
提供病毒颗粒大小、纯度(电泳、不同原理色谱纯度等)、保护性抗原含量、主要蛋白构成及抗原谱分析和完整性等必要的研究资料。
1.2 基因工程重组疫苗
除参照重组治疗用生物制品要求提供适用的相应资料外,对于形成病毒样颗粒的疫苗,还应提供病毒样颗粒关键结构研究的相关资料。如果是纯化的抗原肽或具有保护性特点的表位肽,提供必要的正确性鉴定研究结果。
1.3 病毒载体类疫苗
应对病毒载体类疫苗纯度、序列活性、生物效价、感染性/转导效率、毒力、复制能力、表达目的抗原的正确性等特性进行分析。
1.4 DNA疫苗
应对核酸序列(包括影响疫苗稳定性、转录、翻译表达效率的关键元件)、长度、纯度(超螺旋缺刻、生产过程及贮存期间易出现变化的结构)、生物效价、感染性/转导/转录效率等特性进行分析。
19原料药报产
Q:(云):3类化药的原料药能在其关联制剂申报临床前,单独报产吗?
A:(注册圈):可以提交登记资料关联审评,但不能独立审评。
20持有人变更
Q:(smile):持有人变更,一个品种多个规格,必须一起变更?还有不同规格可以分别转给不同人?
A:(注册圈):如果一个品种多个规格分别有各自的批准证书,则可以分别转给不同人,反之,不可以。
声明:本文转载来源于公众号【注册圈】,所有问答均来源于注册圈社群,非官方问答,仅供参考。
|
|


 |手机版|蒲公英|ouryao|蒲公英
( 京ICP备14042168号-1 ) 增值电信业务经营许可证编号:京B2-20243455 互联网药品信息服务资格证书编号:(京)-非经营性-2024-0033
|手机版|蒲公英|ouryao|蒲公英
( 京ICP备14042168号-1 ) 增值电信业务经营许可证编号:京B2-20243455 互联网药品信息服务资格证书编号:(京)-非经营性-2024-0033






 发表于 2022-12-16 10:58:58
发表于 2022-12-16 10:58:58
 置顶卡
置顶卡 变色卡
变色卡