欢迎您注册蒲公英
您需要 登录 才可以下载或查看,没有帐号?立即注册
x
人工智能(AI)正在彻底改变制药与医疗器械行业的研发、生产与质控方式。随着AI技术的深入应用,相关的法规和指南也加速完善,全球监管格局日益清晰。
- 2023.01.26 美国NIST发布《人工智能风险管理框架》(AI RMF),为组织提供自愿性风险管理流程。
- 2023.12.28 ISO/IEC发布全球首个可认证的AI管理体系国际标准——ISO/IEC 42001,为企业提供系统化应对AI风险的框架。
- 2024.8.01 欧盟《人工智能法案》(AI Act)正式生效,基于风险分级实施强制性监管,高风险AI系统被严格监管。
- 2025.07.07 EU GMP附录22(草案)发布,成为全球首份针对AI的GMP指南,重点聚焦监管静态、确定性输出的AI模型全生命周期管理。
随着EU GMP附录22(人工智能)的推进以及EU AI Act对高风险AI系统的严格监管,提前布局ISO/IEC 42001已成为企业维护合规底线与提升市场竞争力的关键策略。本文结合gempex在GMP领域的丰富经验,为您深入解读ISO/IEC 42001的核心内容,提供精简实用的实施指南。
01 谁应优先实施ISO/IEC 42001?
ISO/IEC 42001适用于所有开发、提供或使用AI系统的组织。对生命科学领域,以下群体尤其适合引入:
- 高风险AI系统开发者:如医疗诊断AI、药物研发预测模型等,需符合EU AI Act第16条对质量管理体系的强制性要求;
- 制药与医疗器械企业:即便AI仅用于非关键环节(如供应链、数据分析),也需通过系统化管理规避 “隐性风险”—— 例如数据溯源缺失导致的结果不可靠,或模型迭代失控引发的合规偏差;
- 产业链供应商:通过ISO/IEC 42001认证可增强客户信任,成为 “合规优先” 的首选合作伙伴。
02 标准核心架构:清晰且易于整合
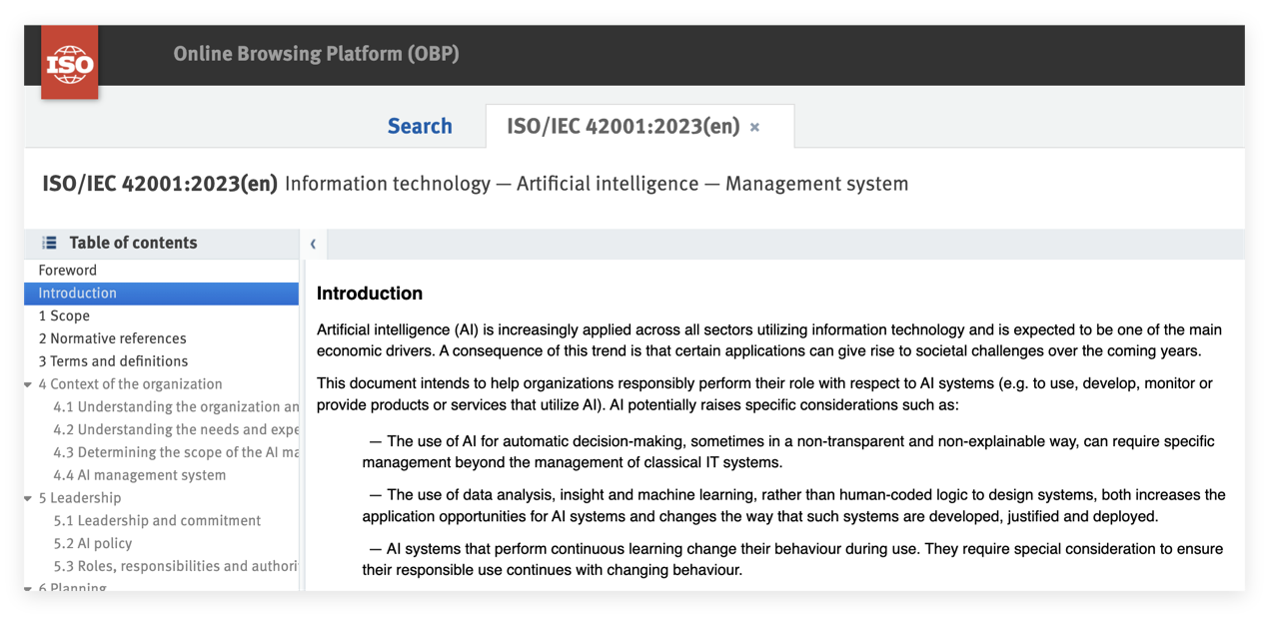
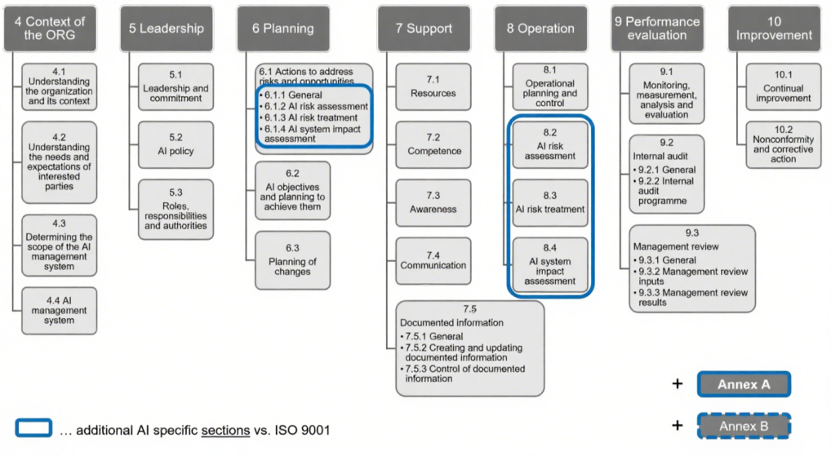
ISO/IEC 42001采用与ISO 9001及ISO 27001一致的高级架构(即“统一章节结构参见图2”), 易于与企业现有管理体系整合。
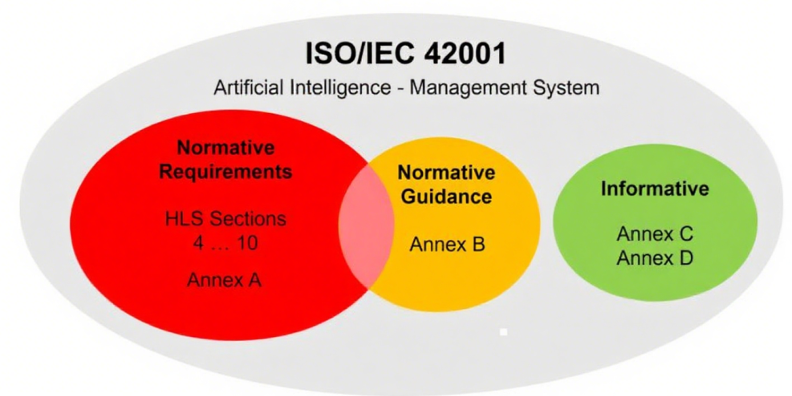
该标准主体内容共11页,其规范性附录是标准的核心组成部分,直接决定体系有效性,需重点关注: 图1:ISO/IEC 42001结构
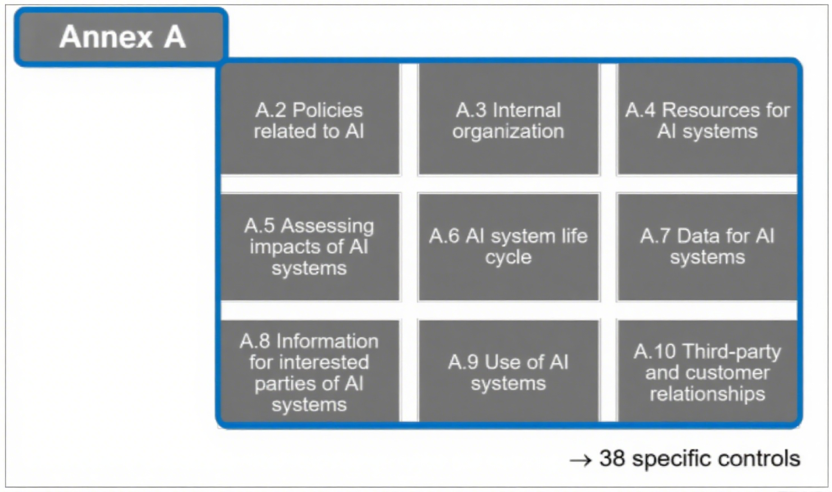
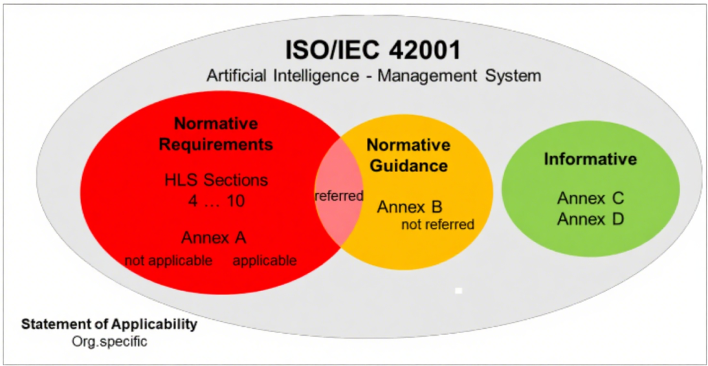
附录A(控制目标与措施):包含38项具体控制要求,包含了数据管理流程实施、数据获取与选择的文档化、数据质量要求界定、AI全生命周期数据溯源及数据处理标准,这些都是ISO 13485未涉及的要求。 图2:ISO/IEC 42001规范性附录A
附录B(规范性实施指南):虽多以“should”等建议性措辞表述,但部分内容被规范性引用,应视为强制要求。企业在适用性声明(SoA)中需说明相关指南的遵循情况。
附录C和D(参考性内容):主要为跨行业应用提供参考,如风险源识别案例,无强制要求,企业可按需借鉴。
图3:ISO/IEC42001‑规范性与参考性附录区别
需特别注意, SoA中必须逐项说明附录A各控制措施的采纳或排除理由,任何未经合理论证的 “豁免” 都可能影响体系有效性。
03 强调风险导向
ISO/IEC 42001以 “风险导向” 为核心,要求风险管理至少包括3个过程:
- AI 风险评估(6.1.2)
- AI 风险处理(6.1.3)
- AI 系统影响评估(6.1.4)
其中,“AI系统影响评估”需分析AI对个人、群体及社会的潜在后果(如医疗 AI 对患者隐私的影响),是风险分析的关键延伸。风险处理需参考附录A中的控制措施,形成与SoA的闭环管理。
标准多处引用ISO/IEC 23894(人工智能风险管理),建议企业将两者结合实施,以满足风险管理的基础性要求。需注意,ISO/IEC 42001对 “风险” 定义为“不确定性的影响”,与 ICH Q9、ISO 13485的定义(“损害发生的概率与严重程度组合”)存在差异,实施时需通过制度设计实现逻辑统一,避免体系冲突。
04 结语
ISO/IEC 42001目前虽非强制,但已代表AI管理领域的先进实践。其核心价值包括:
- 为应对EU AI Act、EU MDR、EU GMP等法规提供系统化路径;
- 对需满足客户高质量要求的供应商,提供了系统化的AI治理框架;
- 对所有组织而言,它能帮助系统化管理AI风险、提升信任度,既保障患者与用户利益,也助力企业可持续发展。
AI领域的竞争不仅是技术迭代的比拼,更是管理体系与治理能力的较量。gempex德恩咨询可基于ISO/IEC 42001为企业提供差距分析、体系整合及认证准备服务,助您在AI创新与合规之间赢得先机。
作者简介 Manuel Goldstein 医疗器械高级顾问 - 拥有14年+医疗器械行业经验,专注GMP合规、质量管理体系及 AI 在医疗领域的应用管控,精通ISO 13485、ISO 9001、MDR、21 CFR 820等法规标准。
- 主导多项关键项目,包括IVD生产商QMS 整改(带领8人团队基于FDA模拟审计推进)、主导组合产品供应商的ISO13485差距分析和医疗设备PLM的 ISO 13485再认证支持等。
- 擅长医疗器械全生命周期管理,涵盖体系监控与维护、风险评估(ISO 14971)、CAPA实施、洁净室及设备确认等。
欢迎垂询 服务热线:400-166-2002
关于gempex德恩咨询 德恩咨询是gempex在中国的全资子公司,是具有国际影响力的GMP咨询与执行机构,致力于为全球的生命科学企业提供合规、高效及可执行的GMP解决方案。经过23年的发展,我们拥有60多位经验丰富的GMP专家,全球累计执行项目超过5000个,累计为1000多个客户提供专业服务,业务遍布20多个国家,并与众多知名药企建立了长期的合作关系。 我们的专家团队拥有丰富的行业经验,熟知NMPA、FDA、EU、WHO、ICH、PIC/S、MHRA、SWISSMEDIC、TGA等GMP法规要求,能为不同国家和地区的客户提供定制化的解决方案,服务包括全球GMP符合性、新厂房合规性、CS计算机化系统、工厂质量管理和多国MAH/MAA服务。
| 

 |手机版|蒲公英|ouryao|蒲公英
( 京ICP备14042168号-1 ) 增值电信业务经营许可证编号:京B2-20243455 互联网药品信息服务资格证书编号:(京)-非经营性-2024-0033
|手机版|蒲公英|ouryao|蒲公英
( 京ICP备14042168号-1 ) 增值电信业务经营许可证编号:京B2-20243455 互联网药品信息服务资格证书编号:(京)-非经营性-2024-0033





 发表于 2025-9-22 17:35:17
发表于 2025-9-22 17:35:17
 置顶卡
置顶卡 变色卡
变色卡