|
5722| 47
|

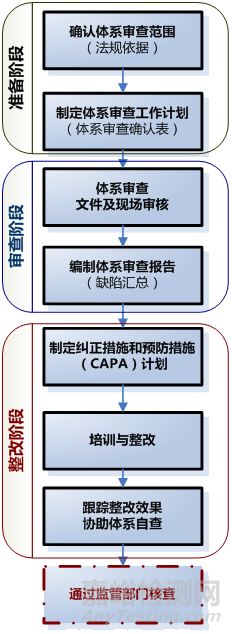
医疗器械生产质量管理体系审查与整改 [复制链接]
[复制链接]
|
 |手机版|蒲公英|ouryao|蒲公英
( 京ICP备14042168号-1 ) 增值电信业务经营许可证编号:京B2-20243455 互联网药品信息服务资格证书编号:(京)-非经营性-2024-0033
|手机版|蒲公英|ouryao|蒲公英
( 京ICP备14042168号-1 ) 增值电信业务经营许可证编号:京B2-20243455 互联网药品信息服务资格证书编号:(京)-非经营性-2024-0033
GMT+8, 2026-4-18 04:14
Powered by Discuz! X3.4
Copyright © 2001-2020, Tencent Cloud.






 发表于 2020-4-27 08:32:33
发表于 2020-4-27 08:32:33

 置顶卡
置顶卡 变色卡
变色卡




