欢迎您注册蒲公英
您需要 登录 才可以下载或查看,没有帐号?立即注册
x
1. Q:各位老师,我想请问国内首次伦理委员会批准日期早于批件日期的原因。是根据哪条法规来的? A1: 伦理前置 Q:是的。就是我不太记得是哪条法规了。因为是要回复这个问题,所以想找一下是哪个法规里规定的 A1: 对伦理前置的,没看到法规的出处,不过看到一段有意思话:2018年7月27日,国家药品监督管理局官网发布《关于调整药物临床试验审评审批程序的公告》(2018年第50号)称,将对药物临床试验审评审批的有关事项作出调整:在我国申报药物临床试验的,自申请受理并缴费之日起60日内,申请人未收到国家食品药品监督管理总局药品审评中心(CDE)否定或质疑意见的,可按照提交的方案开展药物临床试验,这标志着我国临床试验申请默许制正式落地。临床试验审批从“点头制”改成“摇头制”,伦理审查可以在国家局“摇头”之前就进行,这赋予了伦理委员会更大的权利、责任。我院GCP办公室和伦理委员会为了进一步简化了伦理的审批程序,提高伦理审查效率,经过反复讨论与科学论证,最终确定了前置伦理申请与审查流程(见表2)。今后,未取得临床试验批件的国际多中心药物临床试验、一类新药临床试验,均可在等待国家药品监督管理局给出意见的60个工作日期间,到新华医院GCP办公室申请立项和提交伦理委员会进行前置伦理审查。项目负责人除提交常规申请资料外,需提交与国家药品监督管理局的沟通纪要和《前置伦理审评立项资料一致性声明》;对试验方案进行专业性审查后,即可提交资料到伦理委员会进行伦理审查并获得初始审查意见,等待60个工作日后,国家药品监督管理局未给出否定或质疑意见的,即可启动临床试验项目。 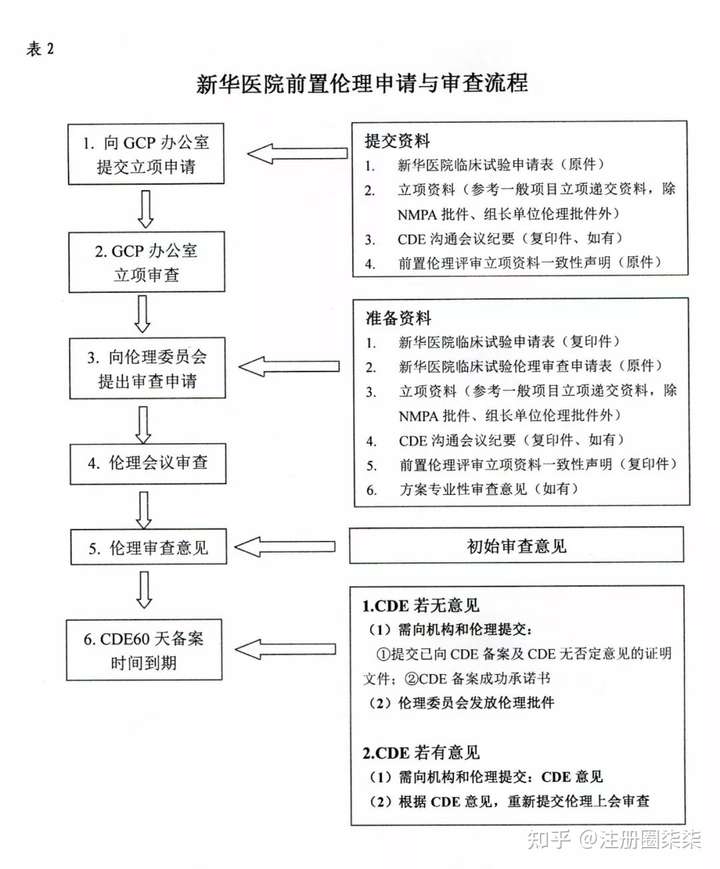 不知道你是需要回复谁提出的问题,我觉得你可以查一下你的试验机构的伦理前置文件 Q: 好的。谢谢。我是在登记临床试验的时候让填的。 应该是最近更新的一项。之前也填过伦理前置的。没让解释原因 A1:那就没办法了,和平台老师解释下吧 Q:嗯嗯。只能这样了
2. Q:请问,现在原料药登记,必须和原研进行杂质对比吗? A1:最好是。其实我个人觉得不对比也可以,药审他们都能找到原研的情况,自己也可以对比到
3. Q:各位老师能分享欧盟法规指南下载网址吗 A1:https://ec.europa.eu/info/index_en
4. Q:想请教一下 申报的产品的批记录和COA还有所有的研究报告上产品名称都是全称,注册的时候产品名称可以使用代码吗 A1:1类新药可以 仿制药不行
5. Q:请问,哪位老师清楚做国际多中心临床,是否需要重新在申请人之窗的临床登记平台登记呢?谢谢 A1:是否用于国内申报 Q:在中国美国都拿到临床批件,目前做国际多中心临床。将来应该中美都要报产。 A1:建议在申请人之窗登记,因为后面在中国报产 Q: 还没登记过国际多中心临床,和登记国内的比,有啥注意事项吗? A2:在中国拿了临床批件就要去做登记,登记平台有填写指南  用 IE浏览器,还需要按照提示安装打印插件 Q: 嗯嗯,之前是以前的RA登记的,以前登记的应该是国内的,现在国内临床变国际多中心临床了,是修改还是重新登记呢? A2:如果两个受理号两个批件,都需要分别登记,只有一个就登记一个就可以了 Q: 是不是原来就有一个国内批件,国际多中心申请又获得一个国内批件?两次申请不是用同一个方案吧? A3: 主要看方案内容、临床阶段、适应症,如原来国内批件做了1期,现在2期是国际多中心。那么就在CDE登记平台新增一项2期的国际多中心临床;同时这个临床要在美国开展,去美国临床登记平台去登记。 要在哪个国家做临床,就去哪个国家登记平台登记(也可以WHO登记成同一个编号),供参考 Q: 非常感谢
6. Q:各位老师,请问模块1中,1.3.1说明书部分,我们是申请注册上市,那是不是1.3.1.1研究药物说明书及修订说明就可以直接忽略了,只放上1.3.1.2上市药品说明书及修订说明就好了呢?还是要给1.3.1.1研究药物说明书及修订说明一个项目封面+不适用说明页? A1:1.3 是按照一个模块放的,1.3.1.1写不适用
7. Q:老师们,中美双报的项目,动物实验是美国做的,在中国报的话试验需要重新做吗?还是翻译成中文就行 A1:不用重新做,翻译就好,安评试验机构有GLP资质
8. Q:各位老师,请教一下:外购原辅料关联制剂,请原辅料厂家出具授权书的时候,各位会有要求在授权书上说明授权期限吗?还是合同上做规定即可?或者不说明即默认长期有效呢?大家实际操作是怎么考虑这个问题的呢?谢谢 A1:我们也是用CDE官方发布的授权书模版,没有授权期限规定,我们的合同也没有特别说明授权期限。 A2:授权书有官方模板,上面没有授权期限的描述
9. Q:各位老师,请教个问题,原料药工厂是否要求具有全检的能力?比如元素杂质订入标准的是用ICP-MS,但是原料药厂家只能用原子吸收法检测这个元素杂质,这种情况是否可以? A1:不可以 Q:谢谢老师,就是说必须用相同方法检测才可以是吧 A1:从质量控制角度来说,这个改变了标准的执行方式,属于中等变更以上,除非获得官方的批准。 Q:那原料药厂家的出厂检验的时候,能不能委托检测? A1:没有绝对的,理论上应当可以。但我没有类似成功的案例。
10. Q:请教一下,我们跟省局申请新药注册二合一符合性检查,省局说查验中心有个书面的检查通知,问了查验中心,查验中心说没有,各位老师有过这样的类似情况吗? A1:那就不用现场检查了,现在检查都是基于风险评估的。
如若有解答不当之处,欢迎大家积极斧正。来源于【注册圈】
| 
 |手机版|蒲公英|ouryao|蒲公英
( 京ICP备14042168号-1 ) 增值电信业务经营许可证编号:京B2-20243455 互联网药品信息服务资格证书编号:(京)-非经营性-2024-0033
|手机版|蒲公英|ouryao|蒲公英
( 京ICP备14042168号-1 ) 增值电信业务经营许可证编号:京B2-20243455 互联网药品信息服务资格证书编号:(京)-非经营性-2024-0033






 发表于 2021-11-15 12:07:59
发表于 2021-11-15 12:07:59

 置顶卡
置顶卡 变色卡
变色卡