金币
UID743052
帖子
主题
积分4789
注册时间2021-9-13
最后登录1970-1-1
听众
性别保密
|
欢迎您注册蒲公英
您需要 登录 才可以下载或查看,没有帐号?立即注册
x
01持有人转让
Q:我们企业不打算生产了,想把持有人跟品种都转移到新厂。现在是计划先转移持有人,获批后新厂再变更生产地址。在持有人转移时,我们需要先把A证变成C,新厂拿B证?还是我们不动,新厂直接拿A证呢?
A1:(感叹号!):转持有人的时候需要先增项。
A2:(苏州 注册 维):老工厂先增C证,新厂拿B证,再转持有人,持有人变更获批后,新厂主导进行场地变更。
A3: 这个情况比较复杂,需要将问题描述清楚。原企业和原生产企业的关系,变更后还有没有关系;等等。
02参比制剂
Q:(南京~注册~小e):上了国家仿制药参比目录,前期买了几支,后面再去买已经不市售了,参比重新备案也无法成功,难道这个品种就不能申报了吗?收录参比制剂目录的那个参比已经不市售了,停产了。但是我们之前买的不够,无法支撑后续的稳定性研究,重新遴选参比备案,也不成功。
A:(杨昌专):要先看下参比不市售的原因,是停产,还是暂时性缺货,或者是安全性的问题。然后再根据参比遴选规则,进行备案。
03一致性评价
Q:(日月光辉):一致性评价批件取得后有没有新旧过渡期?
A:(RA 郭星星):变更管理办法第二十五条。
04稳定性数据
Q:(天津注册如如):现在新药IND申报最少要几个月稳定性数据?6个月是吗?审评期间可以补充稳定性数据吗?
A:(沪-打杂-���):没要求吧,一个月都行。审评期间那得先和主审沟通, 一般pre-ind有ind批一个月稳定性数据就可以去了,然后正式ind也差不多有三个月了。
05参比制剂
Q:(武汉 大蚂蚁):如果单独申报原料药,也是要买参比制剂的吗?
 [backcolor=rgba(18, 18, 18, 0.5)]​ [backcolor=rgba(18, 18, 18, 0.5)]​
编辑切换为居中
添加图片注释,不超过 140 字(可选)
A1:(沪-打杂-���):不买也行。不如买市售份额大的原料药来比。比如二甲双胍你买山东那家的来比。或者让打听打听哪家制剂用了谁家的API过评的 就买那家API来比。
A2:因为原研的原料不易获得,购买参比制剂的目的是为了证明你的原料杂质水平与原研杂质水平没有显著差异。
06一次性进口
Q:(武汉 大蚂蚁):总局关于研制过程中所需研究用对照药品一次性进口有关事宜的公告(2016年第120号)中提及“(四)属于麻醉药品、精神药品、临床试验用生物制品的对照药品,不适用本公告。”,那请问下麻醉药品、精神药品、临床试验用生物制品的对照药品的一次性进口分别参加哪个法规呢?
A1:(京-打雜-���):办事大厅搜搜看,这种得立项吧,生物制品的专门有一个,国家局能搜到,国家药监局关于临床试验用生物制品参照药品一次性进口有关事宜的公告2018 年第 94 号。A2:在网上办事大厅里:麻醉药品研制立项和麻醉药品进口申请项下。
07IND审评
Q:(北京-注册-w): IND申请审评启动后,中间有发补的机会么?
A:(上海 注册 贰雯):没有,就60工作日。
08优先审评
Q:(中美华东注册指间沙):4类的儿童用药,列入了鼓励研发申报儿童药品清单中,可以优先审评吗?A1:(注册圈 苏州 江小白):优先审评需要你preNDA的时候去申请的。按照法规要求,是属于可以申请优先审评的。不过儿童用药一般要新品种,剂型,规格。所以你这个还有点悬,尝试下吧。或者看看在不在鼓励研发申报的儿童用药清单里。
09稳定性考察
Q:(山东 PM Zhu):大家申报固体制剂,开封稳定性一般都做几批?
A1:(沪-打杂-���):2批。
A2:(北京 药物制剂研发 齐越):开封稳定性:影响因素1批;服用期稳定性考察3批。
A3:使用稳定性建议3批。
10联合给药
Q:(上海 RA):如果一个药刚开始是非肿适应症,1期完成了,现在想开一个肿瘤的1期,那么这个1期是否可以直接做联用?
A1:(上海 注册 Frank):这个一期应该需要重新递交IND吧,还需要补充新的药效数据。如果是联用的话,还要根据这个要求,再进行IND申报。我们之前pre-IND和CDE沟通过,需要先拿到单药的PK和安全性数据。前提是创新药。
 [backcolor=rgba(18, 18, 18, 0.5)]​ [backcolor=rgba(18, 18, 18, 0.5)]​
编辑
添加图片注释,不超过 140 字(可选)
 [backcolor=rgba(18, 18, 18, 0.5)]​ [backcolor=rgba(18, 18, 18, 0.5)]​
编辑切换为居中
添加图片注释,不超过 140 字(可选)
A2:(北京 注册 雯):联合给药的前提是获得单药初步安全耐受性数据、DLT剂量和RP2D剂量,如果不同适应症,因为适应症人群不同,耐受程度可能有差异,非肿瘤的I期安全耐受性数据可能不具有代表性。RP2D方面,还要看作用机制,如果作用机制不同,可能在I期研究的标志物或剂量选择的依据不同,可能无法支持联合给药剂量的选择。当然,如果有证据说明上述都是一致的,可以与CDE讨论,是否可以免除单药的I期。我们也没遇到过类似的情况,也只是一点自己的看法,供参考。
11沟通交流会
Q:(w): I期临床试验结束,是需要跟CDE开完I期结束的沟通交流会后才可以开始II期试验吗,还是一边做II期一边准备沟通交流会就可以?
A:(注册圈 苏州 江小白):一期后的沟通交流不是必须的。如果你对二期的方案以及药学,非临床都有信心没问题的话,直接起方案开伦理,伦理过了就可以直接二期了。
12仿制药注册
Q:(中美华东注册指间沙):如果原研进口的在审评中,国内按3类的做了验证性临床可能会早点获批,这个等原研进口获批了仿制的话会有限制吗?
A:(注册圈 苏州 江小白):现在仿制的要求都是一样的。不管有没有国内获批。不会因为你原研进口了要求和不进口相差很大。刚开始引进国内的时候是一样的。只是注册分类不一样。随着国内产品越来越多相应的非临床和临床资料要求会越来越少。你的参比制剂是一样的.
13沟通交流
Q:(广东 化药注册 小榭):如果2期进3期,要跟CDE沟通交流,需要2期总结报告吗?
A:(北京+CRO+周明星):原则上需要交流,这是关键节点!交流需要提交关键数据,不需要盖章的总结报告。
14原料药申报
Q:(东阳光-赵):有人知道前几年受理的没有受理号只有登记号的原料药,如何申请受理号吗?
A:(山东注册君):我们重新申请的单独审评。
Q:(东阳光-赵):是走的这个程序吗?也重新缴费了吧。
 [backcolor=rgba(18, 18, 18, 0.5)]​ [backcolor=rgba(18, 18, 18, 0.5)]​
编辑切换为居中
添加图片注释,不超过 140 字(可选)
A:(山东注册君):我们第一次是I,没有收费,这次是第一次缴费
15生物制品检验
Q:(东阳光-赵):生物制品的临床试验样品,是自检还是必须委托第三方检验?治疗性生物制品。2007版《注册管理办法》:第三十六条 申请人可以按照其拟定的临床试验用样品标准自行检验临床试验用药物,也可以委托本办法确定的药品检验所进行检验;疫苗类制品、血液制品、国家食品药品监督管理局规定的其他生物制品,应当由国家食品药品监督管理局指定的药品检验所进行检验。
A:(YR):治疗性就是都可以。可以自己建立能力的项目自己检,尚未建立的委托检按照方法验证,确认的要求保持方法一致。
16进口药品通关
Q:(Michael苏小川):进口药品通关单时,购货合同的甲方和报验单位不是一家单位,是否可以办理?
 [backcolor=rgba(18, 18, 18, 0.5)]​ [backcolor=rgba(18, 18, 18, 0.5)]​
编辑切换为居中
添加图片注释,不超过 140 字(可选)
A:(南京~注册~小e):可以,但是一次性进口批件的申请单位需要和报验单位一致,甲方和报验单位之间需要有采购委托协议。
17电子签章
Q:(danni):关于电子签章(1)每个pdf文件的首页都要法人和公司的电子签章吗?(2)pdf文件的每页都需要盖公司公章,还是骑缝章?目前的选择是哪种呢?
A1:(yanxinghang):首页公章即可。
A2: pdf文件的每页都需要盖公司公章,申请表、自查表建议有骑缝章。
18变更实施时间
Q:(Piano):《药品上市后变更管理办法》第二十五条 持有人应当在提出变更的补充申请时承诺变更获得批准后的实施时间,实施时间原则上最长不得超过自变更获批之日起6个月,涉及药品安全性变更的事项除外,具体以药品补充申请通知书载明的实施日期为准。这里的“实施时间原则上最长不得超过自变更获批之日起6个月”,是指的批准后6个月内必须要实施新的工艺吗?
A:(扬州注册研发):个人理解,原工艺6个月后不能再生产。
19非临床报告
Q:(注册 竹七):IND或PRE-IND时期的非临床报告,预实验也需要放么?
A:(三迭纪-曹正康-南京-改良型新药):看预实验的试验结果质量如何,是否支持申报。常规性要求是,PIND时候,可以交draft版本的非临床报告或者预实验报告(if available),正式IND时候,仅提交正式试验报告的签字版,PIND期间,毒理试验的部分解剖学结果未出来的,也可以将现有的数据结果整合,先交上去和监管部门沟通。
20临床试验
Q:(刘-注册-武汉):我们公司有个项目,参比制剂原研未进口但是国外已上市,我们属于首仿临床实验会做哪些内容啊?
A1:(sandy):化3,BE和验证性临床,也有小部分是没有开展验证性临床,但是机率很小。
A2:(董):看CDE发的境外已上市境内未上市药品临床技术要求。
Q:(刘-注册-武汉):这个品种是国内已经上市了但是和发布的参比制剂适应症规格不一样,国内临床也都是按超适应症用药的方式在卖。我们想按这个参比制剂的适应症进行三类仿制申报,国内已经有临床应用了,而且原研参比的临床实验数据也可以查得到。这种的能豁免验证性临床吗?
A:(二当家):算是假三类,要具体评估不同适应症和用法用量的安全性的影响。成功和失败的例子都有。具体你这个可以整理好开个沟通交流会。
21补充申请
Q:(硕):增加规格的补充申请,用的是几个月的稳定性数据呢?
A1:(若惜):6个月。
A2:临床期间增加规格,也不一定必然要有6个月稳定性数据,视沟通情况定。
22稳定性数据
Q:(云-原料药注册):核标期间的稳定性数据要怎么提交呢?
A:(左手倒影):在专业审评阶段都可以寄出稳定性资料的,直接寄给CDE。最好给审评老师打个招呼。
23持有人制度
Q:(湖北RA滚雪球):普通公司(非生产型和非研发机构)能做持有人吗?普通口服制剂。
A:(江苏-注册-果酿):可以。药品管理法:药品上市许可持有人(以下简称持有人)是指取得药品注册证书的企业或者药品研制机构等。
 [backcolor=rgba(18, 18, 18, 0.5)]​ [backcolor=rgba(18, 18, 18, 0.5)]​
编辑
添加图片注释,不超过 140 字(可选)
24稳定性研究
Q:(上海-注册-V):仿制药稳定性研究 溶曲的拟合始于自己的0天拟合还是与参比制剂拟合?
A:(勤劳的小蜜蜂):参比制剂。
Q:(上海-注册-V):请问是0天还是稳定性试验中(6,12个月等)参比制剂?
A:(勤劳的小蜜蜂):我们做同一时间跟参比制剂拟合。
25有效期查询
Q:(Chloe):参比制剂说明书找不到有效期,还有其他方法查有效期吗?
A:(景元):查下参比制剂的审评报告或者电子说明书,应该有有效期。
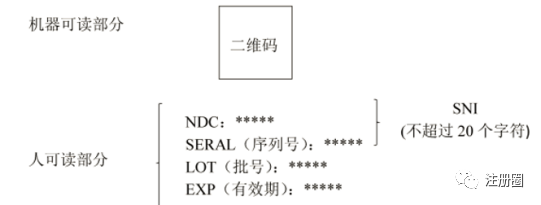
Q:(Chloe):FDA没有,也没查到在其他国家上市,原研以前在国内上市的说明书和仿制药的说明书都是2年,感觉就是2年了。
A:(京-GMP-David):按照美国的要求,包装要标的,识别码。
 [backcolor=rgba(18, 18, 18, 0.5)]​ [backcolor=rgba(18, 18, 18, 0.5)]​
编辑
添加图片注释,不超过 140 字(可选)
Q:(Chloe):确实有。不过说明书上是空白的,或许可以问问卖参比的。
A:(京-GMP-David):说明书上应该是没强制,所以你看盒子和瓶子,按照法案要求,是要有标识码和条码,标识码是那个供应链安全法案提出来的,条码是21CFR201里的要求。
26注册批偏差
Q:(陈嘉音):注册批可以开偏差吗?是否注册批一旦有偏差就不能算成功的注册批了
A:(京-GMP-David):看偏差严重程度,除非是对产品质量有很大影响甚至导致批次失败的。
Q:(陈嘉音):如果注册批开了偏差,是不是要在注册递交之前关掉偏差才能算不影响到注册?
A:(京-GMP-David):这个跟产品放行有关系,放行前要审核偏差情况的,个别的偏差可能需要持续跟踪。
27原辅包登记
Q:(冷了个冷):我是进口已上市品种,原辅包部分未登记。由于产品已上市多年,没法追溯原资料。对于已上市品种,未登记的原辅包还需要重新要求供应商登记吗?
A:未登记的原辅包需要重新要求供应商登记。
28研究者手册
Q:(Arti):研究者手册是基于活性成分写的是吗?IND申报期间,不同适应症一份研究者手册么?
A:研究者手册是基于活性成分也要结合适应症,不同的适应症需要不同的研究者手册。“《药物临床试验质量管理规范》第三十四条 申办者提供研究者手册,其内容包括试验药物的化学、药学、毒理学、药理学和临床的(包括以前的和正在进行的试验)资料和数据。”当同一个化合物不同的作用靶点和作用机制对应不同的适应症时,临床研究者手册中出化学、药学外,其他内容都可能不一致,无法共用。
29自制对照品
Q:(安徽注册王宁):CEP注册,杂质对照品想要自制的话,需要准备哪些证明资料吗?A:提供自制对照品制备信息,结构确证的证明。
30通用名命名
Q:细胞产品药品通用名称命名是提交药典委吗?还是INN呢?
A:先去WHO申请英文的通用名,然后药典委会根据英文的给中文的。
31注册检验
Q:个体化的抗肿瘤细胞治疗产品在申请BLA之前,标准复核和样品检验是由中检院进行吗?如果是的话,需要申请前置注册检验吗?
A:由中检院进行,可以申请前置注册检验。
32IND申报
Q:关于细胞治疗产品,如果IND申报了单适应症,后续其他适应症申报IND时,能不能直接上II期呢?药学连续3批生产、长期稳定性研究、药理毒理研究还需要重新以新适应症患者来源的细胞做吗?
A:如果I期研究数据充分可以直接上II期;需要。
33电子签章
Q:(L Z L):按照1月1日CDE的新要求提交电子光盘,申请表如果按照1.2版本的电子签章软件盖章,只能盖一处章,也不能盖骑缝,另外MAH和生产企业不是一家公司的话,就得盖2个章,用1.2版本的软件就没法实现了。有同行最近提交资料成功受理的吗?关于申请表电子签章这块是怎么操作的?
A1:软件不是非常成熟,1.2版本不能加盖骑缝章,建议打CDE受理电话咨询。
A2:IND期间,仅盖了自己公司的章,未签委托生产机构章,无补正受理了。
34复方制剂申报
Q:(赵小飞):中药和化药组成的复方制剂,应该按照什么报?中药1.1复方制剂吗?
A:如果是该复方制剂为已上市制剂,可以考虑化药4类;如果是有已知活性成份的新复方制剂,且具有明显临床优势;可以考虑化药2.3类 。如果处方未在国家药品标准、药品注册标准及国家中医药主管部门发布的《古代经典名方目录》中收载,具有临床价值,且未在境外上市的中药新处方制剂,可以按1.1。
35方法验证
Q:(丫头):异构体采用自身对照法定量,方法验证做线性时,主成分的线性范围是按多少做的啊?会做到1%的150%或200%?
A:定量限到1%的150%或200%。
36小规格产品申报
Q:(上海-RA-Astoria):大规格药品审评期间,小规格产品可以通过新的上市许可申请或补充申请申报吗?还是需要等大规格产品获批后,再通过补充申请进行申报呀?
A:可以通过新的上市许可申请;也可以等大规格产品获批后,再通过补充申请进行申报。
37新增适应症
Q:(上海-注册-M):新药临床试验期间想要增加适应症,是按补充申请提交资料1-8吗?都需要准备哪部分资料?
A:获准开展药物临床试验的药物拟增加适应症(或者功能主治)以及增加与其他药物联合用药的,申请人应当提出新的药物临床试验申请,经批准后方可开展新的药物临床试验。按现行版新药临床试验申请要求提交资料。
38BE豁免
Q:(广州-化学仿制药-茜茜):关于仿制药栓剂生物等效性豁免的问题:有没有做了大规格栓剂生物等效性,小规格栓剂BE豁免的呢?或者有指导相关指导原则和法规的吗?
A:对于局部给药全身作用的制剂,应进行生物等效性研究;可以参考《 以药动学参数为终点评价指标的化学药物仿制药人体生物等效性研究技术指导原则》进行风险评估。
声明:本文转载来源于公众号【注册圈】,文章版权归原作者所有,所有问答均来源于注册圈社群,非官方问答,仅供参考,如若了解更多信息可自行前往查看。违规请版主删除。
|
|


 |手机版|蒲公英|ouryao|蒲公英
( 京ICP备14042168号-1 ) 增值电信业务经营许可证编号:京B2-20243455 互联网药品信息服务资格证书编号:(京)-非经营性-2024-0033
|手机版|蒲公英|ouryao|蒲公英
( 京ICP备14042168号-1 ) 增值电信业务经营许可证编号:京B2-20243455 互联网药品信息服务资格证书编号:(京)-非经营性-2024-0033






 发表于 2023-2-10 09:50:28
发表于 2023-2-10 09:50:28
 置顶卡
置顶卡 变色卡
变色卡