金币
UID744339
帖子
主题
积分1519
注册时间2021-9-16
最后登录1970-1-1
听众
性别保密
|
欢迎您注册蒲公英
您需要 登录 才可以下载或查看,没有帐号?立即注册
x

01 IND概述与分类
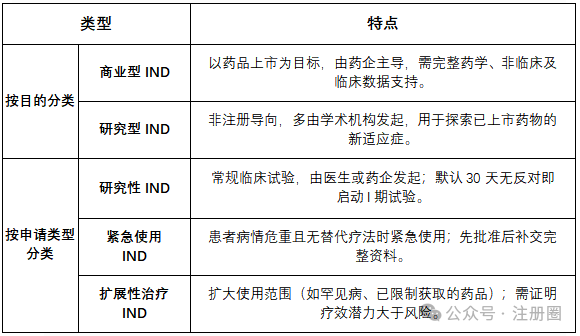
IND(Investigational New Drug)即新药临床试验申请,是药物从实验室研究迈向人体试验的关键节点。其核心目的是向美国FDA证明候选药物的安全性合理性,确保临床试验受试者风险可控。根据FDA规定,任何未获批药物在美国进行跨州运输或临床试验均需通过IND申请。近年来随着基因治疗、免疫疗法等新兴领域的崛起,IND申请持续呈现多元化增长趋势。IND申请包含多种类型,具体见下表:
 [backcolor=rgba(0, 0, 0, 0.1)]
[backcolor=rgba(0, 0, 0, 0.1)] [backcolor=rgba(0, 0, 0, 0.1)]
[backcolor=rgba(0, 0, 0, 0.1)]
添加图片注释,不超过 140 字(可选)
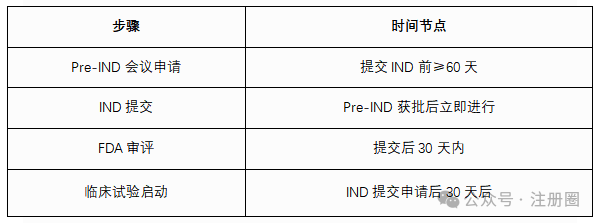
02 IND申请全流程
 [backcolor=rgba(0, 0, 0, 0.1)]
[backcolor=rgba(0, 0, 0, 0.1)] [backcolor=rgba(0, 0, 0, 0.1)]
[backcolor=rgba(0, 0, 0, 0.1)]
添加图片注释,不超过 140 字(可选)
1. Pre-IND会议
1.1 申请提交
- 申办方需通过FDA电子提交网关(ESG)或邮件提交正式申请,包括产品名称、适应症、会议类型(B类)、议程及问题清单等。
- 申请需在会议召开前至少60天提交,FDA通常在21个自然日内回复是否批准。
1.2 资料准备
申办方需在会议前30天提交约50-100页的简报文件,内容包括:◆ 化学、制造和控制(CMC)信息(生产工艺、质量标准等);
◆ 非临床研究(药理毒理、药代动力学);
◆ 临床开发计划(I期方案设计、适应症背景等);
◆ 拟讨论问题的背景及数据支持。
1.3 会议召开
- FDA在收到申请后60天内安排会议(书面回复、面对面、电话或视频形式),通常仅允许同一药物在IND阶段申请一次Pre-IND会议。
- 会议时长约1小时,申办方可就非临床研究设计、CMC要求、临床方案等核心问题提问,FDA提供即时反馈。
- FDA在会议后30天内提供正式纪要,记录讨论要点及共识,申办方可提交补充记录以澄清理解偏差。
1.4 会议内容
- 非临床研究:动物毒理设计、药效学/药代动力学数据是否支持人体试验、安全起始剂量推算等。
- CMC要求:生产工艺、初步质量标准、非常规辅料或病毒清除研究(如重组蛋白)。
- 临床方案设计:I期剂量选择、纳入/排除标准、终点指标、儿科研究计划等。
- 监管策略:数据呈现方式、IND申请资料清单及潜在风险控制措施。
1.5 注意事项
- 会议价值:可减少冗余研究、避免临床暂停,缩短上市时间。
- 替代方案:若申请过早(如早期毒理阶段),可考虑INTERACT会议(更早期概念验证沟通)。
- 文件要求:若会议后需补充数据,应明确问题范围,避免多次申请。
2. IND申请提交
2.1 资料准备
按CTD格式整合药学、非临床及临床方案(详见第三部分)。
2.2 提交方式:
- 电子提交:通过FDA的电子提交网关(ESG)提交,格式为PDF或特定电子格式(如XML)。
- 纸质提交(仅限紧急情况):需提前与FDA协商。
3. FDA审评流程
3.1 IND审评目标
- 确认药物安全性足以支持首次人体试验(FIH)。
- 评估数据完整性、科学合理性及合规性(如GMP、GLP)。
3.2 审评阶段(30天内)
- 形式审查:
FDA检查申请资料是否完整,确认申请文件的完整性及基础合规性。
- 实质性审评:
◆ CMC审评:关注生产稳定性、杂质控制是否符合cGMP要求。
◆ 非临床审评:评估动物实验数据能否外推至人类(如NOAEL→安全起始剂量)。
◆ 临床审评:审查临床试验设计的科学性(如剂量选择、风险控制措施)。
3.3 审评结果
- 批准(Approve):允许开展临床试验,通常附带条件(如额外监测)。
- 不批准(Refuse):列出缺陷,申办方根据要求提交修正方案。
- 无行动(No Action):默认批准,若FDA未在时限内提出异议。
03IND申报资料内容与格式
a. 提交IND的要求
(1) 封面页(表格FDA-1571)
申请人须提交包含以下内容的IND封面页:(i) 申请人名称、地址、电话号码,申请日期及研究药物的名称;(ii) 拟开展的临床研究阶段;(iii) 承诺在IND生效前不启动临床研究;(iv) 承诺由机构审查委员会(IRB)负责研究的初始审查和持续监督,并由研究者向IRB报告研究方案的变更;(v) 承诺遵守其他适用的法规要求;(vi) 监督临床研究进展的负责人姓名及职务;(vii) 负责评估药物安全性信息的人员姓名及职务;(viii) 若申请人将部分研究职责转移至合同研究组织(CRO),需声明CRO名称、研究标识及转移的职责范围(若全部职责已转移,可提交概括性说明);(ix) 申请人或其授权代表的签名。若签字人不在美国境内,需提供在美国境内的公证代理人或授权官员的姓名、地址及联署签名。
(2)目录
(3)引言与总体研究计划
(i) 简要说明药物名称、活性成分、药理类别、已知结构式、剂型、给药途径、研究目标及拟开展研究的总体时长;
(ii) 总结药物在人体中的既往经验(包括其他国家的研究或上市情况,若与安全性相关);
(iii) 若药物因安全性或有效性问题在任一国家被撤市,需列明国家及原因;(iv) 拟开展研究的年度计划概要,包括:
◆ 研究依据与目标;
◆ 拟研究的适应症;
◆ 研究方法概述;
◆ 首年计划开展的临床试验类型;
◆ 预计受试者人数;
◆ 基于动物毒理或人体研究预期的严重风险。
(5)研究者手册(如适用)
(i) 原料药与制剂的描述,包括结构式(若已知);(ii) 药物在动物和人体中的药理作用及毒理效应总结;(iii) 药代动力学与生物利用度总结;(iv) 人体临床研究的安全性与有效性信息摘要(可附已发表文献);
(v) 基于既往经验的潜在风险、副作用及研究期间需采取的预防措施或特殊监测。
(6)研究方案
(i) 每项计划的试验方案。未在IND中首次提交的方案,应根据法规要求后续提交。
◆Ⅰ期研究:方案可较为简略灵活,需包括受试者估算数、安全性排除标准及剂量计划(如最大剂量、给药方法),并明确关键安全监测指标(如生命体征、血液生化);
◆ Ⅱ期与Ⅲ期研究:需提交详细方案,设计中应包含替代方案或应急措施(如早期转换疗法)。
(ii) 方案须包含以下要素(具体内容视研究阶段而定):(a) 研究目的;(b) 研究者资质(简历或资格证明)、协作研究者及研究机构信息;(c) 受试者入选/排除标准及人数估算;(d) 研究设计(如对照组设置、减少偏倚的措施);(e) 剂量确定方法、最大剂量及暴露时长;(f) 观察指标与测量方法;(g) 临床操作、实验室检测或其他风险控制措施。
(7)化学、制造与质量控制(CMC)信息
(i) 根据研究阶段提交原料药与制剂的组成、生产工艺及质量控制信息。早期阶段侧重原料与原料药的识别与控制,原料药和制剂的最终标准需在研究结束时确定;
(ii) 稳定性数据要求随研究阶段和时长调整。短期研究需相应有限的稳定性数据;(iii) 随着生产规模扩大,需提交补充信息以支持更大范围的试验;(iv) 具体要求包括:
原料药:理化特性、生产工艺、质量控制标准及稳定性数据;
制剂:成分清单(包括辅料)、生产工艺、质量控制标准及稳定性数据;
安慰剂:成分、制备与包装说明;
标签:提供给研究者的所有标签副本;
环境评估:根据法规要求提交豁免声明或环境评估报告。
(8)药理与毒理学信息
需提供动物或体外研究中药物的药理作用、作用机制、吸收、分布、代谢、排泄及毒性数据,以支持拟开展临床试验的安全性结论。(i) 药理学与药物代谢动力学;(ii) 毒理学:
◆ 急性、亚急性和慢性毒性试验结果;
◆ 生殖毒性及发育毒性数据;
◆ 与给药途径相关的特殊毒性(如吸入、皮肤或眼部);
◆ 体外毒性研究;
◆ 符合GLP规范的声明(若适用)。
(9)既往人体经验(i) 若药物曾在美国或其他国家开展研究或上市,需提交相关安全性及有效性信息(尤其与拟研究适应症相关的受控试验数据);(ii) 若为复方药物,需分别提交各活性成分的信息,除非某一成分已获FDA批准或合法上市;(iii) 若药物曾在美国以外国家上市,需列明上市国家及因安全性或有效性问题撤市的国家及原因。
(10)附加信息(i) 药物依赖性:若药物具有精神活性或滥用潜力,需提交相关研究数据;(ii) 放射性药物:需提供辐射剂量计算数据;(iii) 儿科研究:儿童安全性与有效性评估计划;(iv) 其他信息:可能影响安全性评估或试验设计的补充说明。
(11)其他需提交的信息若FDA要求,需提供其他相关资料以供审评。
b. 已提交信息的处理
申请人无需重复提交既往提交的内容,但可通过引用归档文件(注明文件名、编号、卷册及页码)。引用第三方提交的信息需获得授权声明。
c. 外文材料的翻译要求
所有非英文部分需提交准确完整的英文翻译,并附原文出版物副本。
d. 文件份数
需提交原件1份及副本2份(包括修订件及报告)。
e. IND编号规则
所有IND相关提交需按顺序使用三位连续编号(初始IND为000,后续修订、报告等依次递增)。
f. 知情同意例外情况标识
若研究涉及知情同意豁免,需在封面页显著标注。
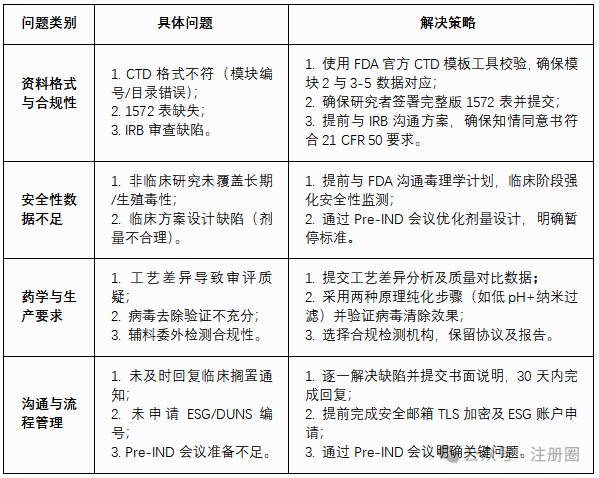
04常见申报问题与解决策略
 [backcolor=rgba(0, 0, 0, 0.1)]
[backcolor=rgba(0, 0, 0, 0.1)] [backcolor=rgba(0, 0, 0, 0.1)]
[backcolor=rgba(0, 0, 0, 0.1)]
添加图片注释,不超过 140 字(可选)
05IND申报未来趋势
5.1 技术驱动下的申报效率提升
☆ AI与自动化工具的应用
◆ 数据整合与分析:通过机器学习优化非临床数据(如毒理研究)与临床数据的关联性分析,缩短审评周期;
◆ 文档自动化生成:利用自然语言处理技术(NLP)自动生成CTD格式文件,减少人工错误并提升模块间一致性;
◆ 风险预测模型:基于历史审评数据构建算法模型,预判FDA关注点并提前优化申报策略。
☆ 真实世界证据(RWE)的整合FDA逐步接受RWE作为传统临床试验数据的补充,例如:
- 利用电子健康记录(EHR)验证药物在特殊人群中的安全性;
- 结合可穿戴设备数据优化剂量递增设计。
5.2 政策与监管框架的革新
☆ 审评流程的持续优化
- 默示许可制的扩展:细胞治疗等领域的IND审评时间已缩短至60日,未来可能推广至更多治疗领域;
- 动态风险评估机制:针对低风险药物(如已上市的食品/化妆品用于新适应症)豁免部分IND要求,仅保留核心安全性审查。
☆ 国际化协同与双轨申报
- ICH标准深化:通过ICH成员国互认机制,缩短中美欧三地IND时间差;
- 审评数据互认:FDA与NMPA等机构探索非临床研究数据的跨境互认,减少重复试验。
06结语
IND申报是新药研发的“通关文牒”,其成功取决于科学严谨性与策略性沟通。企业需精准把握法规要求、模块化撰写资料,并善用FDA的Pre-IND会议机制,才能高效推进管线。未来,随着技术革新,IND申报将面临更复杂的挑战,持续关注FDA指南更新、灵活运用数字化工具与全球化策略将成为制胜关键。
参考文献
[1] 21 CFR Part 312 Investigational New Drug Application
[2] 《联邦食品、药品和化妆品法案》(FD&C Act)
[3] ICH M4(R4): Organization of the Common Technical Document for the Registration of Pharmaceuticals for Human Use
声明:本文转载来源于公众号【注册圈】,文章版权归原作者所有,如若了解更多信息可自行前往查看。违规请版主删除。
更多资料请前往注册圈网站
注册圈 杭州几度数据技术有限公司
|
|


 |手机版|蒲公英|ouryao|蒲公英
( 京ICP备14042168号-1 ) 增值电信业务经营许可证编号:京B2-20243455 互联网药品信息服务资格证书编号:(京)-非经营性-2024-0033
|手机版|蒲公英|ouryao|蒲公英
( 京ICP备14042168号-1 ) 增值电信业务经营许可证编号:京B2-20243455 互联网药品信息服务资格证书编号:(京)-非经营性-2024-0033





 发表于
发表于  置顶卡
置顶卡 变色卡
变色卡
